

Bioinformatics
Introduction and practical applications
Course TFTB29
|
|
Bioinformatics Course TFTB29 |
| |
|||
|
Please submit your answers via the ARMS system http://bioinfo.ifm.liu.se/edu/ARMS/TFTB29/HT2013/report_assignment_3.html |
|||
Sequence informationIntroductionDomains are the "building blocks" of proteins. They are regions that fold independently and are often interconnected by flexible linker regions. In general, each domain is associated with a distinct function, for example hydrophobic membrane-spanning domains, cofactor-binding domains, and catalytic domains. To study relationships between proteins, a common first step is to compare the amino acid sequences using a multiple sequence alignment (MSA). A small example is shown here: 
From the MSA it is possible to determine the conserved regions in the proteins. Proteins often contain clusters of residues that are conserved because of particular requirements on their interactions, either internally in the protein or with the environment. Usually, these residues are of functional importance to the protein, for example for the binding properties or the catalytic activity. The function and structure of proteins can thus be characterized by their conserved sequence motifs, and this can be automated using various computational methods, e.g. machine learning techniques. In this assignment we will first get acquainted with patterns, which is the most basic method for sequence motif recognition. Then we will expand our view to the more advanced profiles, after which we will move on to the powerful statistical hidden Markov models (HMM), which represent one of the most sensitive classification methods that exist today. Finally, we will look at some tools for calculating some physical properties of proteins. AssignmentProsite is a well used resource that contains a database of patterns and profiles and also provides web based tools that allows users to analyze proteins online. Use the tools in Prosite to scan the protein RON_HUMAN. 3-1. Does your protein have any known domains? If so, which? PatternsA pattern is usually a number of consecutive residues important for a specific biological function. These regions include binding sites or enzymatic catalytic sites. Here is one example:
[AG]-x-C-x(4)-{DE}
This pattern is translated as: Ala or Gly, any, Cys, any, any, any, any, anything but Glu or Asp. 3-2. Which pattern matches the ATP binding region of RON_HUMAN? Give both the ID and the actual pattern! Hint: To see the details for a pattern, click the link next to the pattern ID (on the form PS00000). Now, we're going to go into even more detail for this pattern, so once again click on the link next to the pattern ID (should have the exact same text as the one you just clicked, but will not bring you to the same page). If everything went OK, you should see a page that contains lots and lots of identifiers (further down). 3-3. How good is this pattern? For swissprot, give the number of correct hits (true positives), wrong hits (false positives) and missed sequences (false negatives). Consider the following two hypothetical patterns that describe the same conserved region: [FW]-C-x(3)-C-[AG]-E-[MLI]-D and [FW]-C-x(3)-C-[ASGPT]-E-[IVLM]-D 3-4. Which of the patterns is more tolerant (less restrictive)? Motivate your answer! 3-5. What are the consequences of using a more tolerant pattern in searches?
OK, lets play some more with patterns! Open this rudementary pattern test utility (Hint: open in a new window or tab)!
Construct the following patterns and answer with the most restrictive (least tolerant) pattern possible.
3-7. Extend the pattern region by three amino acids to the right and one to the left (for example in Prot3 that would correspond to the following region: G-S-S-V-T-N-V-K-V). All but Prot2 should be included in the match!
3-8. Finally, update your pattern so that the corresponding region of Prot2 will also be included! (The matched region in the MSA should not change from 3-7) ProfilesProfiles are more sensitive and more robust than patterns and can therefore be used to describe larger sequence features, such as for example domains. Profiles are also called position specific scoring matrices (PSSM), and as the name suggests, they consist of matrices of numbers which are used to score sequences based on which amino acid residue they have where. Now, getting Prosite to show details on profiles is a bit trickier. Do as you did for the pattern details and go back to the Prosite scan summary and bring up the documentation page for the first matching domain. On this page, once again click on the link next to the profile ID (should once again have the exact same text as the one you just clicked, but will not bring you to the same page). If everything went OK, you should now be looking at a page with lots and lots of numbers on it. Further information regarding profiles can be found through the Prosite help pages, among other places here. 3-9. Given that "/M" means match and "/I" means insertion, give a plausible description as to what the algorithm does when it matches a sequence to this profile! 3-10. Given an optimal match between a profile and a sequence, what are the hit criteria? (i.e. what does it take for the algorithm to call it a hit?) Are there any differences compared to the pattern case, and if so, how does this affect the reliability of the results? 3-11. Profiles are made from MSAs. Give a plausible explanation to how this is done! Use the example MSA at the top of the page! (briefly and conceptually, max 100 words) Hidden Markov models (HMM)HMMs are in fact very similar to profiles. Both have match, insert and delete states, and both have specific ranking of amino acid residue types for individual positions. However, while profiles are an empirical attempt to generalize BLAST-like alignment scoring to individual scoring of each alignment position, HMMs have a much more solid base in mathematical statistics and are therefore more reliable. The figure below shows a very simple HMM, which is very short and also uses DNA (simply because DNA has fewer letters than proteins). 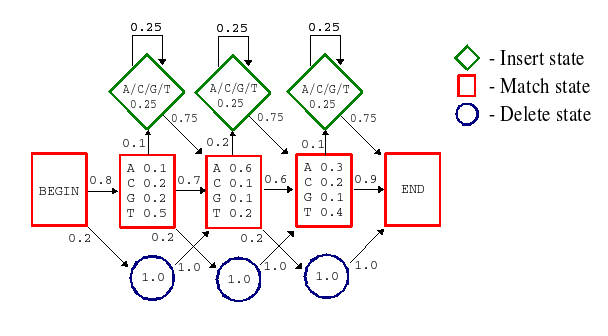
In HMMs, everything is a statistically computed probability. The profile scores have been replaced by probabilities; the general insertion and deletion penalties from profiles have been replaced by specific transition probabilities between states; there are probability distributions for the types of amino acid residue present in insertions, and so on. All this makes the HMMs very sensitive and powerful, and they can therefore be used to model very large and widely diverse groups of sequences, such as for example protein superfamilies. A less simplified illustration of an HMM (matching a protein family) is available here: Less simplified HMM example [SAM drawmodel format, pdf]. As a comparison, figure 2 below shows a simplified example of how a profile can be represented by a HMM. 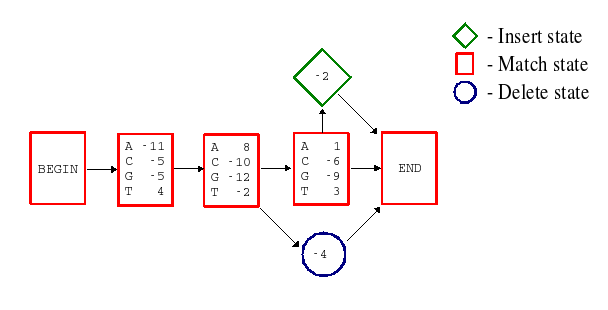
3-12.
Describe how the DNA sequence TAGT could be matched to the HMM in figure 1 (Not figure 2 above but the one with probabilities!).
Give also the resulting probability of the optimal match!
3-13. How do you think a HMM distinguish a sequence matching from a sequence not matching the HMM? Are there any differences compared to the profile case? 3-14. HMMs are made from MSAs. Give a plausible explanation to how this is done! Use the example MSA at the top of the page! (briefly and conceptually, max 100 words) Protein familiesPfam has a database of high quality HMMs that can be used to reliably characterize proteins. Use the Pfam web interface to scan RON_HUMAN for matching HMMs. Note: In addition to its own HMMs, Pfam also uses external tools to classify the query protein (e.g. SMART, seg, signalp, etc). For this assignment, however, we will ignore all hits that do not come from Pfam itself. 3-15. Pfam has two sections, PfamA and PfamB. Which is more reliable? Explain why! 3-16. Does Pfam find any sequence features, predicted by PfamA, that Prosite did not? If so, which? 3-17. Did Prosite find any sequence features that Pfam does not, given PfamA predictions only? If so, which? Clicking on a Pfam hit will take you to the corresponding documentation page, which holds both biological and technical information on the hit. For your amusement, you can click on the link "[Download HMM]" far down to the right to see what a HMM really looks like on the inside. 3-18. Using Prosite and Pfam, try to find as much information on RON_HUMAN as possible. What do you think this protein does in the cell? Which parts do what? Please, give your best theories on the protein's function, localisation, interactions, etc... One way of studying protein similarities is to search for homologues in sequence databases, using for example BLAST. Another way is to use databases that contain information on sequence patterns and protein family conservation, such as Prosite and Pfam. 3-19. Which are the advantages and disadvantages of using such sequence pattern databases compared to using databases of amino acid sequences and BLAST? 3-20. If patterns, profiles and HMMs are essentially the same thing (made from the same kind of data, used for the same purpose, ...), how come the older methods are still in use? Why not pick the most advanced and sensitive method and use that for everything? Now, we will take a closer look at domains as building blocks for proteins. A mosaic, or chimeric, protein is partly built of domains or segments which are similar or identical to domains of other proteins. The process involved is called exon shuffling. The mosaic protein is created when an exon from one gene is integrated into another gene's intron. Another kind of exon shuffling is when an exon is duplicated in the same gene. 3-21. If exon shuffling has occurred, what is required to get a functional protein? Give at least two requirements.
Consider the following proteins: 3-22. First, use any tool or resource to find out what these protins do in the cell, and give an explanation in layman's terms!
Now, compare the proteins in Pfam. 3-23. Which of the proteins is obviously chimeric? Note that they may all be chimeric, but one of them is clearly built of domains from all the others. This is the text book example of a chimeric protein. 3-24. Restricting your theories to only these four proteins, which is the most likely ancestral protein for each domain in the chimeric protein? Other toolsHere is the primary structure for a naturally occurring fusion protein. >Fusion protein MEPAPARSPRPQQDPARPQEPTMPPPETPSEGRQPSPSPSPTERAPASEE EFQFLRCQQCQAEAKCPKLLPCLHTLCSGCLEASGMQCPICQAPWPLGAD TPALDNVFFESLQRRLSVYRQIVDAQAVCTRCKESADFWCFECEQLLCAK CFEAHQWFLKHEARPLAELRNQSVREFLDGTRKTNNIFCSNPNHRTPTLT SIYCRGCSKPLCCSCALLDSSHSELKCDISAEIQQRQEELDAMTQALQEQ DSAFGAVHAQMHAAVGQLGRARAETEELIRERVRQVVAHVRAQERELLEA VDARYQRDYEEMASRLGRLDAVLQRIRTGSALVQRMKCYASDQEVLDMHG FLRQALCRLRQEEPQSLQAAVRTDGFDEFKVRLQDLSSCITQGKAIETQS SSSEEIVPSPPSPPPLPRIYKPCFVCQDKSSGYHYGVSACEGCKGFFRRS IQKNMVYTCHRDKNCIINKVTRNRCQYCRLQKCFEVGMSKESVRNDRNKK KKEVPKPECSESYTLTPEVGELIEKVRKAHQETFPALCQLGKYTTNNSSE QRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAAC LDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQ LLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRK RRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLE NSEGLDTLSGQPGGGGRDGGGLAPPPGSCSPSLSPSSNRSSPATHSP 3-25. Where has the fusion occurred? Give the residue number! 3-26. What is the function and subcellular location of the proteins that have generated this fusion protein? 3-27. What domains do these proteins contain? 3-28. What disease is the fusion protein associated with? At the Expasy web site, there is a general plot tool, ProtScale, that given a Swissprot sequence ID or an amino acid sequence makes plots of various properties (selectable by the user) against the amino acid sequence. You shall now test this tool in order to construct a hydrophobicity plot according to Kyte and Doolittle on the human 5-hydroxytryptamine 1A receptor (Swissprot ID 5HT1A_HUMAN). Use window size 15, which is appropriate when searching for transmembrane regions. You will se a limited number of broad peaks with values above 1.5, corresponding to the transmembrane regions. 3-29. How many such peaks do you see? Signal sequences, that direct newly synthesised protein to its final localisation where it should fulfill its function, are often encoded in the N-terminal parts. There are now several predictors available for such predictions. One good site is the Center for Biological Sequences in Copenhagen -- www.cbs.dtu.dk. In this exercise, you shall use some of their predictors in order to predict the presence or absence of signal sequences and organelle localisation for three sequences below with given Swissprot IDs. (Thus, you first need to get the amino acid sequence using e.g. SRS.)
NOTE: We will not use the new version 4.0 of SignalP as the update makes predictions more unreliable for these particular questions. SignalP 3.0 and Target P can be found here :
Use SignalP to predict the signal sequences and
TargetP to predict the organelle localisation for
the following sequences with given Swissprot ID:s. Detailed information about
interpretation of the results is available via the web pages. Note that two models
are employed by SingalP and that you may have to consider both results!
3-30. What signalpeptide and organelle location (if any) do you find for USHA_ECOLI? 3-31. What signalpeptide and organelle location (if any) do you find for AMPL1_SOLLC? 3-32. What signalpeptide and organelle location (if any) do you find for ALDR_BOVIN? Links
Pfam
http://pfam.sanger.ac.uk/ (search) http://pfam.sanger.ac.uk/help (help)
Prosite
http://www.expasy.ch/prosite/ (search) http://www.expasy.ch/prosite/prosuser.html (user manual)
ProtScale
http://www.expasy.ch/cgi-bin/protscale.pl
SignalP and TargetP
http://www.cbs.dtu.dk/services
|
|||
|
|
|||
|
Please submit your answers via the ARMS system http://bioinfo.ifm.liu.se/edu/ARMS/TFTB29/HT2013/report_assignment_3.html Check your marking process at http://bioinfo.ifm.liu.se/edu/ARMS/TFTB29/HT2013/check_progress.html |
|||
Problems? |
|||
|
|||
Modified
October 2011 |
|||